English
English
For millions of people worldwide,3,4 understanding sickle cell disease is more than an academic exercise—it is a way of life. While most people who carry the trait do not have serious health complications, certain physically stressful conditions may cause complications such as muscle breakdown5. When two parents carry the trait, there is a 25% chance that each of their children will be born with sickle cell disease. And for those people who have the disease, it can mean a life filled with exhaustion punctuated by severe pain.6
Sickle Cell Statistics
Although conditions with similar symptoms had been described earlier,7 the sickle cell anomaly was first published in Western medical literature by James Herrick,8 describing the blood of a severely anemic dental student from Granada, West Indies. A decade later, sickle cell was identified as the first genetic disease.9,10 And while the genetic mutation that leads to sickle cell disease occurred several thousand years ago,11 the global burden of the disease is only now being realized.
Sickle cell disease is a global public health problem. More than 300,000 babies are born with sickle cell disease each year, and many of them die before the age of five.1,12 Worldwide, 0.1% of children are born with Sickle Cell Disease, but the prevalence varies by region and ethnicity. People of African descent make up 90% of those carrying at least one sickle cell gene—the other 10% are those with Hispanic, South Asian, Southern European, and Middle Eastern ancestry.13,14 In some parts of Africa, up to 45% of the population is known to carry the trait.1,15 In the United States, one in every 13 Black children is born with the sickle cell trait, and one in every 365 Black children is born with sickle cell disease.2 For Hispanic Americans, sickle cell disease occurs in one of 16,300 births. 4
What is Sickle Cell Disease?
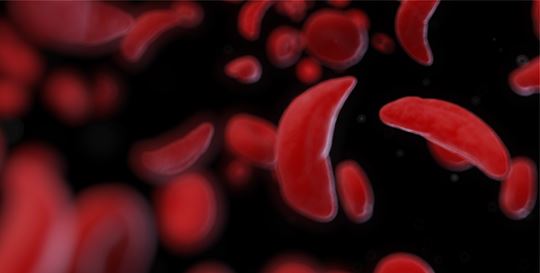
Sickle cell disease is a group of autosomal recessive inherited blood disorders caused by single nucleotide mutation in the gene encoding the hemoglobin protein. Sickle cell disease can’t be “caught.” People are either born with the mutation or not. Normal hemoglobin, which carries oxygen throughout the body, forms a tetramer containing two beta and two alpha subunits. Sickle cell hemoglobin (Hemoglobin S (HbS)) has a mutation in the beta subunit. In the most common form of sickle cell disease, individuals inherit a copy of the mutant HbS from each parent (HbSS). When the body has sufficient oxygen, HbS-carrying red blood cells (RBCs) look like normal RBCs—biconcave, smooth, round discs. But when the cells are low in oxygen, the HbS molecules become insoluble, causing them to polymerize into long, rigid chains that push up against the cell membrane and change the cell’s shape to the classic sickled (crescent) shape.
Sickled cells are rigid and fragile, which shortens their lifespan. Regular RBCs live for about 120 days.16 The lifespan of a sickled RBC is 80% shorter—only about 2 weeks.17 The spleen, responsible for removing old and damaged RBCs from the bloodstream, is more likely to remove sickled cells, resulting in the anemia associated with sickle cell disease. Lower RBC levels mean lower blood oxygen, which puts further oxygen stress on the body, leading to more sickling of the cells. The crescent-shaped RBCs are stickier than their normal counterparts. They can stick to and damage the blood vessel walls, activating the clotting response, blocking the blood vessels, and preventing oxygen from getting to vital organs. Individuals who are homozygous for HbS can have significant acute pain from both low oxygen and clot formation, and are at higher risk for stroke, heart disease, kidney disease, vision loss, and infection.18
So why do harmful genes such as the sickle cell trait persist in the population? Interestingly, heterozygous carriers naturally protect against malaria, a blood-borne parasite replicating in RBCs. The link between the sickle cell trait and protection against malaria was first suggested in the mid-1900s.19,20 Individuals living with the sickle cell trait show a significantly lower parasite load compared with those without the trait, and children with the trait clear the parasites faster. Polymerization of the HbS slows the growth of the parasite.21 Since malaria persists, the sickle cell trait persists.
Diagnosing and Monitoring Sickle Cell Disease
Early sickle cell diagnosis and prompt treatment of crisis events are essential for survival.14,15,22 A complete blood count with differential (CBC-Diff) may be used to aid in tracking sickle cell disease and monitoring treatment.23,24 Levels of RBCs and reticulocytes may be used to determine RBC turnover and monitor the hemolytic anemia associated with the disease. Red cell distribution width (RDW) may be measured to monitor different RBC populations.
Patients receiving hydroxyurea therapy may need further monitoring. Mean corpuscular volume (MCV) may be measured to track the desired increase in size caused by the drug.23,25,26 Conversely, hydroxyurea can cause neutrophil levels to get dangerously low, meaning that patients may not be able to fight off infection,27 so measuring white blood cells (WBCs), especially neutrophils, is necessary. Neutrophils must also be measured in patients not receiving hydroxyurea therapy, as high levels may indicate infection.
Even with advances in automated blood cell analysis, a peripheral blood smear may be helpful for the differential diagnosis of sickle cell disease.28
For the millions of people living with sickle cell disease, there is hope. Blood transfusions can help improve the oxygen debt. Medications such as hydroxyurea and L-glutamine have been developed to help with pain or decrease the likelihood of HbS polymerization. Research continues to find new and better medications to help mitigate symptoms. Most recently, bone marrow transplants,6 CRISPR-Cas 9 gene editing, and gene therapy are employed to alter patient genetics permanently.29 And increased education from people like the Colemans, who speak out to destigmatize and bring awareness to the disease.
Learn more about the Complete Blood Count on Beckman Coulter’s hematological systems and the Full-Field Peripheral Blood Smear Application® on the Scopio X100 and X100HT.